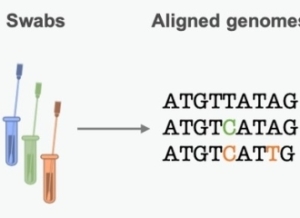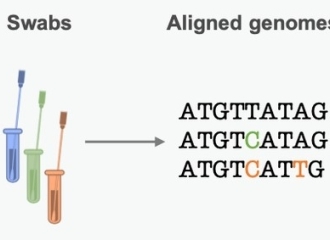

图1种系发生树是基于基因序列变异对病原体过去传播链的估计。(图片翻译:swabs鼻拭子,aligned genomes比对序列,phylogenetics trees种系发生树)来源:原文
全球已有超过2.5亿个体新冠检测阳性,大多是通过鼻拭子检测。阴性的样本检测完就会当做生物废弃物处理掉,但检测出阳性的样本会保留下来,为科学家们提供关于新冠病毒的宝贵信息,从中可能发现有关疫情的新特点。
借助谱系动力学,研究者们可以追溯病原体的基因变化史,从而推断疫情爆发的时间地点、病原体传播方式以及是否存在未被发现的感染者。谱系动力学同样能够帮助我们了解和追溯新变种的传播,奥密克戎变种就是一个典型例子。
鼻拭子里的玄机
病原体和人一样都有属于自己的基因组。基因组是该生物所有遗传信息的总和,它对于生物的新陈代谢和复制繁衍来说都是必需的。
如今,测序一个病原体的基因组耗时更少、成本更低。在瑞士,一个由政府资助的科学院已经从近8万份新冠阳性鼻拭子标本中提取了病毒的基因组。
科学家们将从不同患者的样本中获得的基因序列进行比对以发现不同之处。而这些差异就是病毒基因组复制过程中发生的小错误——突变(mutation)。他们可以通过这些突变来复现传播链,进而推算出当前疫情的动态趋势。
谱系动力学:拼接变异线索的工具
现在研究者得以从谱系动力学的角度来描述不同突变对当前疫情走向造成的影响。通过这种方法,研究者可以从原始数据获取病毒或细菌基因组突变的位置并进一步了解这些突变带来的影响。这听起来貌似有点复杂,但想直观地了解原理其实非常简单。
病原体在人际间传播时会发生突变。随着疫情演变,病原体会累积大量突变。科学家们会使用种系发生树(phylogenetic tree)来直观概括突变病原体之间的关系,就像病原体的“家谱”一样。每个分支点代表了病原体的一次传播,即它从一个个体传播到了另一个个体。
分支长度与测序样本之间的差异大小成正比。短的分支意味着分支(即变异)产生的时间间隔很短,说明病原体传播的速度很快。研究分支长度可以帮助我们了解病原体过去的传播速度,甚至在病原体传播早期、我们尚未发现疫情时的传播情况也能了解。

图2病原体基因组测序数据可用于构建种系发生树并推测疫情动态趋势。分支越短,传播越快。(图片翻译:outbreak origin:传播起点,first detected case:首例病例,speed of spread:传播速度在不断提升,time:时间)来源:原文
用数学模型描述疾病动力学
一般来说,模型是对现实的简化表示。最常用的模型便是数学模型。我们能在很多地方看到研究者们用数学方程来描述现实生活中的某个核心过程。在谱系动力学中,科学家们用方程描述了种系发生树和疫情进展之间的关系。
以结核病为例,它是一种最致命的细菌感染。如今由于抗生素滥用,耐药的结核杆菌越来越多,感染后也越来越难治愈。一位耐药结核病病人的治疗时间可达数年之久。为了预测耐药结核杆菌未来带来的疾病负担,我们来估算一下它的传播速度。

图3流行病学家致力于追溯病原体传播到另一个种群的过程。(图片翻译:transmission传播,uninfected person未感染个体,infected person感染个体,treatment治疗,cured person康复个体,resistance development出现耐药性,death死亡,antibiotic-susceptible bacteria对抗生素敏感细菌,antibiotic-resistant bacteria耐药细菌)来源:原文
为了实现这一目标,我们要对疾病传播的两个关键过程即感染和获得耐药性过程建立数学模型。现实生活中,感染者会传播病原体给他人,同时他们也会得到治疗,最后要么康复,要么不幸病死。除此之外,在这个过程中耐药病原体也会越来越多。

图4谱系动力学模型将现实生活中流行病发展的过程简化为数学方程和参数来帮助研究者们研究。(图片翻译:transmission rate:传播率,patients infected with antibiotic-susceptible bacteria:个体感染了对抗生素敏感的病原体,cure/death rate:治愈/病死率,resistance development rate:出现耐药性率,patients infected with antibiotic-resistant bacteria:个体感染了对抗生素耐药的病原体)来源:原文
下面,我们将上述流行病发展的几个过程转化为一个数学模型。在这个模型中有两组病人,第一组感染了对抗生素敏感结核杆菌,第二组感染了对抗生素耐药结核杆菌。流行病学发展的重要过程,如病菌传播、患者治愈或病死,在两组的发生率不同。一旦结核杆菌出现耐药性,那么相应患者即从第一组移到第二组。
尽管这个模型忽略了结核病流行过程中的某些因素(如无症状感染或治疗后复发),但在应用到种系中不同结核杆菌基因组时,仍然有助于科学家估算耐药结核病的传播速度。
流行病背后的秘密
谱系动力学方法可以帮助研究者回答一些仅靠确诊病例无法全面回答的问题,比如未检测到的病例数量是多少,或者新一轮疫情发生的源头在哪。
我们最近在欧洲对高致病性禽流感病毒(highly pathogenic avian influenza,HPAI)H5N8所做的调查研究便是这种基因组研究的典型案例。H5N8病毒在2016年掀起了一波疫情,席卷了30个欧洲国家,家禽和野生鸟类都未能幸免。最终,数千万家禽被捕杀,家禽行业遭受重创。
但是这场疫情的传播者究竟是家禽还是野生鸟类?显然鸟儿是无法告诉我们答案的。而我们从家禽农场和野生鸟类栖息地取样搜集到了H5N8病毒的基因组。通过进行谱系动力学建模,我们找到了答案:在某些国家疫情主要是通过家畜农场之间传播造成的,而在另外一些国家则是野生鸟类将病毒带到了家禽农场造成的。
在这个例子中,我们帮助不同国家的畜牧兽医部门根据各国特点来制定针对性防疫政策。这意味着某些国家可能要限制家畜农场之间的流通,另一些国家则要注意限制野生鸟类与家畜之间的接触。
现在,我们运用谱系动力学分析来评估新冠疫情防控政策(如尽早封闭边境和封城)的效果。它的一个显著优势在于可以计算出未被检测的病例。这个模型甚至可以模拟疫情早期缺乏样本时病毒传播的大致情况。
谱系动力学模型正在迅速发展,应用领域越来越广,数据集的规模也越来越大。然而,如何将基因组测序工作扩展到未采样物种和未采样地区,如何保持公共数据快速分享,这些依旧是该方法面临的挑战。相信未来有一天,这些数据和模型将会帮助人们以一个新颖的视角了解流行病,更好地进行疾病防控。注:该文章发表于2021年12月1日,其中数据的时效性或许已发生变化。
撰文:Claire Guinat;Etthel Windels;Sarah Nadeau,他们是苏黎世联邦理工学院,进化计算博士后。
翻译:孙正杰
审校:安君
引进来源:Theconversation

本文来自:中国数字科技馆
特别声明:本文转载仅仅是出于科普传播信息的需要,并不意味着代表本网站观点或证实其内容的真实性;如其他媒体、网站或个人从本网站转载使用,须保留本网站注明的“来源”,并自负版权等法律责任;作者如果不希望被转载或其它相关事宜,请与我们接洽。
[责任编辑:环球科学]